编者按:基础研究以科学的方法和标准来研究和评价疾病的病因,确定和评价疾病的诊断方法、防治措施的效果和效益,可使临床医学得到不断发展和进步。医学离不开基础研究,与临床实践是互为推动的关系。在本届AHA年会上,亦有最新基础研究(Late-Breaking Basic Science,LBS)的公布,现将其中一个专题的重要发现呈现给广大读者。
MAVS对硫化氢诱导的心肌缺血损伤具有保护效应
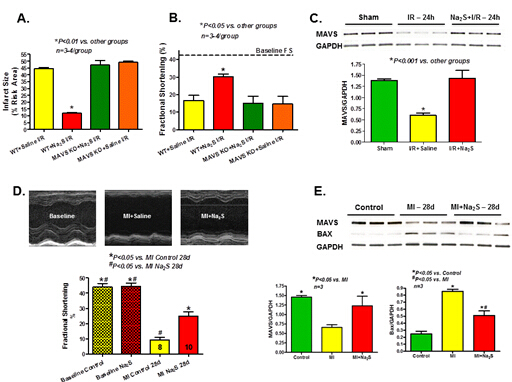
既往研究显示,硫化氢(H2S)可部分通过保存线粒体完整性发挥对心肌梗死(MI)及炎性反应的保护效应。鉴于线粒体抗病毒信号(MAVS)蛋白可缓解氧化应激或缺血所致Bax介导的细胞色素C自线粒体释放,弗吉尼亚联邦大学David Durrant等的研究旨在确定MAVS是否参与了H2S的心脏保护作用。
在基线行超声心动图检查后,通过结扎冠状动脉30分钟使野生型成年小鼠及MAVS基因敲除小鼠发生MI,再松开结扎冠状动脉行再灌注24小时。MI前1小时应用Na2S(100 μg/kg,腹腔注射)或生理盐水对受试小鼠预处理。结果发现,在野生型小鼠中,与生理盐水预处理小鼠相比,Na2S预处理小鼠MI后24小时TTC染色所测梗死面积更小,左室短轴缩短率(FS)更高;但MAVS基因敲除小鼠中则未见此现象(图A和图B)。此外,各组小鼠的风险区并无差异。Western blot分析显示,MI后24小时心肌MAVS表达显著降低,而采用Na2S预处理可使MAVS表达恢复(图C)。对另一部分小鼠行永久冠状动脉结扎,并采用Na2S或生理盐水干预28天。结果发现,生理盐水对照组小鼠MI后28天时LVFS显著降低,而Na2S治疗可使LVFS显著恢复(图D)。此外,与生理盐水对照组相比,Na2S预处理组小鼠三色染色法所测左室梗死面积更小(22.4%±2.7% vs. 33.5% ±2.1%,P<0.05),生存率更高(P<0.05)。Western blot分析显示,MI后28天时MAVS表达显著下降,且MAVS表达与Bax表达有相关性(图E),而Na2S可显著抑制上述变化。
可见,Na2S对MI有保护作用,可预防MI诱导的心衰发生,MAVS可能在其上述效应中发挥一定作用。保留MAVS及补充H2S有望成为缺血性心衰治疗手段。
硫氧还蛋白-2可抑制线粒体ROS生成及Ask1活性从而维持心功能
硫氧还蛋白-2(Trx2)是可通过抑制线粒体ROS生成、阻断凋亡信号调节激酶(Ask1)依赖的凋亡信号从而调节细胞氧化还原及存活的关键线粒体蛋白。耶鲁大学Wang Min等研究发现,与健康非衰竭心脏相比,在扩张型心肌病(DCM)患者心脏中,随着氧化应激标志物的增高以及ASK1磷酸化/活性的增加,Trx2蛋白表达水平下降。心肌特异性Trx2敲除小鼠(Trx2-cKO小鼠)可在1月龄时出现自发DCM,其心脏体积增大、室壁厚度降低及左室收缩功能进行性下降,最终可致小鼠在4月龄左右死亡。此外,Trx2-cKO小鼠心功能的下降会伴随线粒体超微结构破坏、线粒体膜去极化、线粒体ROS生成增多及ATP生成减少,而这些均与ASK1信号上调及心肌细胞凋亡增多有关。应用高选择性、小分子ASK1抑制剂可显著减轻Trx2-cKO小鼠的氧化应激、凋亡、纤维化及心衰,减轻左室扩张,改善左室功能并降低其死亡率。对Trx2基因缺陷心肌细胞研究显示,抑制ASK1可减轻细胞凋亡,减少线粒体ROS生成。综上可见,ASK1是Trx2调控心肌细胞的重要靶点,通过基因敲除Trx2激活ASK1可导致心功能不全。该研究表明,线粒体Trx2可通过抑制线粒体ROS生成及ASK1依赖的细胞凋亡在保护心功能中发挥至关重要的作用。
支链氨基酸分解代谢缺陷可导致葡萄糖代谢受损并加重心肌缺血/再灌注损伤
支链氨基酸(BCAA)即亮氨酸、异亮氨酸和缬氨酸是维持人体蛋白质平衡、能量平衡及营养信号转导所必需的氨基酸。其在线粒体中的代谢受支链酮酸脱氢酶(BCKDH)复合体调控。线粒体局部的磷酸酶2C(PP2Cm)是调节BCKDH活性的关键酶。既往研究发现,PP2Cm缺陷可导致BCAA分解代谢缺陷,氧化应激增加,并可导致斑马鱼发生心功能不全。华盛顿大学Zhen Zhang等对PP2Cm基因敲除小鼠研究发现,与野生型小鼠相比,其2个月时超声心动图评估的心功能正常(FS:44%±3% vs. 43%±2%,P=NS),离体灌注心脏中的心肌高能磷酸含量及31P 核磁共振波谱所测心肌等容收缩功能也正常。但13C核磁共振同位素分析显示,与野生型小鼠相比,基因敲除小鼠心脏中葡萄糖氧化减弱(16%±3% vs. 26%±2%,P=0.018),脂肪酸氧化增加(51%±4% vs. 39%±3%,P=0.020),糖原含量可降低超过50%(4.4±0.5 vs. 10.9±1.8 ?mol/g,P=0.000)。尽管与野生型小鼠相比,基因敲除小鼠基因敲除2个月时血糖及胰岛素水平正常,但6个月时可发生高血糖(空腹血糖水平:128±36 vs. 77±6 mg/dl,P=0.036)及高胰岛素血症(血清胰岛素水平:0.68±0.33 vs. 0.22±0.02 ng/ml,P=0.037)。上述结果表明,PP2Cm基因敲除小鼠存在葡萄糖代谢受损。进一步研究发现,低灌流缺血25分钟后再灌注40分钟可使野生型小鼠的心功能恢复至51%±11%,但却仅能使基因敲除小鼠的心功能恢复至8%±3%(P=0.002)。此外,基因敲除小鼠灌注期间心脏中磷酸肌酸、ATP及无机磷的水平无法恢复至野生型小鼠水平。过表达胰岛素依赖性葡萄糖转运体GLUT1可增加基因敲除小鼠对葡萄糖的摄取与利用,从而减轻或延缓缺血/再灌注损伤,心功能可恢复至49%±9%。总之,该研究结果表明,BCAA分解代谢缺陷可导致葡萄糖代谢受损,进而引发胰岛素抵抗、加剧缺血/再灌注损伤。
在1例癫痫发作时不明原因猝死患者中发现的SCN8A突变在小鼠模型中可导致心肌兴奋性改变
电压门控钠离子通道(VGSCs)突变与癫痫性脑病发病相关,存在该突变人群在癫痫时发生不明原因猝死(SUDEP)风险增加。目前,SUDEP发病机制尚不清楚,越来越多的证据表明,自主神经和/或心肌细胞中VGSC突变所致心律失常可能在其中发挥了非常重要的作用。密歇根大学Chad R Frasier研究小组在1例合并SUDEP的癫痫性脑病患者中发现了一种新生突变即SCN8A-N1768D。在异源系统中对野生型小鼠及突变Nav1.6小鼠钠电流对比分析发现,该突变可使神经元中瞬态及持续性钠电流密度增加。除了神经元外,Nav1.6在心脏中也有表达,且优先表达于T管系统,可诱发河豚毒素敏感型(TTX-S)钠电流。为探讨SCN8A-N1768D突变是否会改变心肌兴奋性,研究者创建了SCN8A-N1768D/+(D/+)小鼠。对急性分离的新心室肌细胞电流钳检查发现,与野生型小鼠心室肌细胞相比,D/+小鼠心室肌细胞延迟后除极的发生率显著增加(4/5 vs. 0/5),动作电位早期复极相延长(APD:30% vs. 50%),但总动作电位时程、动作电位上升支幅度及峰值均无显著变化。这与TTX-S VGSC对心室肌细胞总钠电流影响较小一致。体表心电图显示,与对照小鼠相比,D/+小鼠心率有减弱趋势(P=0.06)。腹腔注射2 mg/kg去甲肾上腺素后,上述心率差异消失。腹腔注射120 mg/kg咖啡因后,D/+可出现与室性心动过速相似的心电图表现,野生型小鼠则不会出现这种情况。但离体心脏标本的突变小鼠心率不再减慢,提示研究所观察到的心动过缓可能与副交感神经活性增加有关。综上可见,SCN8A在心脏兴奋性的维持中发挥了重要作用,其基因突变不仅会导致癫痫,还会导致心律失常,从而导致SUDEP。









 京公网安备 11010502033353号
京公网安备 11010502033353号